
Evidenzgenerierung – ein Schwerpunkt der MDR.
Wir zeigen Ihnen, wie Sie sich in 7 Schritten eine gute Ausgangssituation verschaffen.
Market
Access
08.04.2018
Noch viel Zeit bis zum Startschuss? – eher nein!
Die EU-MDR, mit dem Ziel entworfen Sicherheit und eine schnelle Versorgung der Patienten mit innovativen Medizinprodukten zu gewährleisten, trat am 25. Mai 2017 in Kraft und beinhaltet eine dreijährige Übergangsfrist. Während dieser Übergangszeit kann wahlweise entweder altes (Richtlinie 93/42/EWG; sowie 90/385/EWG) oder bereits neues Medizinprodukterecht angewendet werden.
Ab dem 26. Mai 2020 ist die neue EU-Medizinprodukte-Verordnung dann verpflichtend anzuwenden, weshalb es sehr ratsam ist, bereits jetzt schon, in der Übergangsphase Erfahrungen mit der MDR zu sammeln. Damit dies gelingen kann, müssen allerdings auch die Voraussetzungen auf Seiten der Zertifizierungsstellen, der sogenannten Benannten Stellen, erfüllt sein.
Verbände sehen bereits heut schon Engpässe beim Aufbau der neuen Benannten Stellen, da diese viele neue Pflichten und einen großen Bedarf an neuen Fachkräften (bspw. klinische Experten, Biometriker u.a.) haben. Es wird daher erwartet, dass die ersten MDR-Zertifizierungen voraussichtlich ab Sommer 2019 starten können (vgl. Notified Body Operations Group (NBOG)). Aus diesem Grund sieht die MDR vor, dass Zertifikate, die von Benannten Stellen nach dem alten Medizinprodukterecht ab dem 25. Mai 2017 ausgestellt werden, ihre Gültigkeit auch über den 26. Mai 2020 hinaus behalten, längstens jedoch bis zum 27. Mai 2024 (vgl. offizielle MDR- und MDD-Texte).
Strategisch betrachtet wäre daher eine Re-Zertifizierung Ende 2019 / Anfang 2020 zielführend, wenn man möglichst lang nach altem Recht resp. den darin enthaltenden Anforderungen zertifiziert sein möchte. Doch täuschen Sie sich nicht, denn bereits heut wird es schwer insb. für kleinere Unternehmen einen Termin bei einer Benannten Stelle zu erhalten und der Andrang Mitte 2019 wird groß werden. Sollten Sie zudem bisher nur sehr rudimentär klinische Bewertungen durchgeführt haben, werden Sie den Zeitraum auch hierfür benötigen, um sich gut für die Zeit nach 2020 aufzustellen.
Auch die MDD forderte Evidenznachweise – was ist nun anders?
Kurz zusammengefasst: die MDR fordert zum einen umfangreichere Informationen zum anderen konkretisiert sie diese auch im Vergleich zur MDD. Die folgende Tabelle gibt Ihnen einen auszugsweisen Überblick über die zeitaufwendig und erfolgskritischen Kriterien, die im Falle von klinischen Prüfungen auch teuer werden können:
| Kriterium | MDD | MDR |
|
Anforderungen in der klinischen Bewertung (Kapitel 6 der MDR) |
Ziel der klinischen Bewertung sehr vage: Leistungen müssen dem Leistungsziel des Produkts entsprechen und unerwünschte Nebenwirkungen ermitteln. Anhang X (Art. 1) |
Wesentlich höhere Anforderungen in der klinischen Bewertung: der Hersteller muss im Rahmen der Nutzen-Risiko Bewertung nachweisen, dass die verwendeten Daten belastbar und zuverlässig
sind. Eine ausführliche Überprüfung der Fachliteratur ist erforderlich. Alle verfügbaren relevanten klinischen Daten sowie sämtliche Lücken im klinischen Nachweis müssen ermittelt und
analysiert werden. Art. 61 (1, 2, 3), Anhang XIV (Teil A) |
|
Methodische Qualität der klinischen Prüfungen (Kapitel 6 der MDR) |
Die methodische Qualität klinischer Prüfungen ist nicht näher spezifiziert, z.B. "angemessenen Prüfplan". Art. 15, Anhang X (Art. 2) |
Die methodische Qualität der klinischen Prüfung wird unter Einbeziehung des Studiendesigns, des statistischen Ansatzes und weiterer methodischen Aspekte kritisch überprüft. Wenn
klinische Nachweise nicht ausreichend sind, muss der Hersteller zur Beantwortung offener Fragen neue oder zusätzliche klinische Daten erzeugen. Art. 70, Anhang XV |
|
Ethische Überprüfung der klinischen Prüfungen (Kapitel 6 der MDR) |
Ethische Überprüfung sehr kurz: Ethik nach Helsinki Erklärung für jeden Schritt der klinischen Prüfung (Studienidee bis Veröffentlichung). Anhang X (Art. 2.2) |
Die ethische Überprüfung ist sehr ausführlich darzulegen. Diese erfolgt durch eine Ethik-Kommission gemäß dem nationalen Recht. Allgemein gilt ein besserer Schutz von Patienten, die
an klinischen Studien teilnehmen. Es erfordert bspw. eine ausführliche Darlegung der Einwilligung der Aufklärung bei klinischen Prüfungen für unterschiedliche Patientengruppen. Art. 62 (3), 63-76 |
|
Meldefristen in der Marktüberwachung (Kapitel 7 der MDR) |
Marktüberwachung sehr kurz dargestellt: Hersteller hält ordnungsgemäße Verpflichtungen aus dem Qualitätssicherungssystem ein. (Art. 10) |
Kürzere Meldefristen in der Marktüberwachung: Hersteller müssen regelmäßig aktualisierte Berichte über die Sicherheit von Produkten der Klassen IIa (bei Bedarf bzw. mindestens alle 2
Jahre) und der Klassen IIb und III (mindestens einmal im Jahr) erstellen. Art. 86 |
|
Klinische Überwachung (Kapitel 7 der MDR) |
Strengere klinische Überwachung nachdem Produkte auf den Markt gebracht wurden. Art. 93, Anhang XIV (Teil B) |
|
|
Audits und Produktprüfungen (Kapitel 7 der MDR) |
Angekündigte und nicht-angekündigte Audits und Produktprüfungen. Art. 93 (3) |
|
|
Zusätzliche PMS Berichte (Kapitel 7 der MDR) |
Hersteller müssen zusätzliche Berichte, z.B. Post Market Surveillance Pläne und Berichte mit Nutzen-Risiko-Abwägung erstellen. Art. 84, 85, 86, 88 |
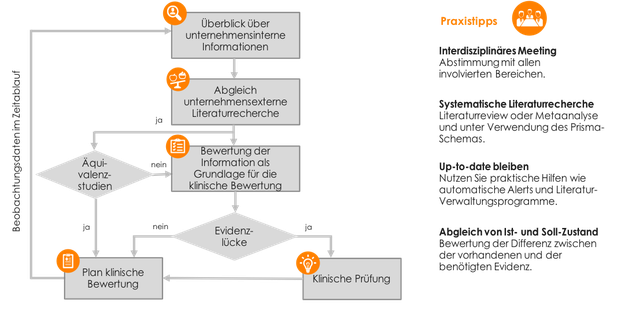
Vor dem Hintergrund der oben aufgeführten Neuerungen, lassen sich die Aufgaben grob wie folgt einteilen und in Abbildung 1 zusammenfassen:
A. Klinische Bewertung beinhaltet die systematische Recherche der wissenschaftlichen Fachliteratur. Die ermittelten relevanten klinischen Daten und Lücken im klinischen Nachweis müssen ausgewertet und analysiert werden. Dabei dürfen nur Daten zu einem technisch, biologisch und klinisch vergleichbaren Produkt berücksichtigt werden. Technische Vergleichbarkeit bedeutet ähnliche Bauart und Anwendungsbedingungen, biologische - Verwendung gleicher Materialien und klinische - der gleiche klinische Verwendungszweck.
B. Klinische Prüfung dient der Vervollständigung der klinischen Bewertung. Sie beschreibt eine systematische Untersuchung des Medizinprodukts, wenn im Rahmen der klinischen Bewertung festgestellt wurde, dass die vorhandenen Daten nicht ausreichend sind. Zu solchen Prüfungen gehören randomisierte kontrollierte Studien (RCT) und müssen wissenschaftlichen, ethischen und methodischen Standards entsprechen. Beispielsweise muss die getestete Population der Zielpopulation entsprechen und der klinische Verwendungszweck klar erkennbar sein. Solche Prüfungen sind (bis auf wenige Ausnahmen) Pflicht für implantierbare Produkte und Produkte der Risikoklasse III (Art. 61 (4) MDR).
C. Post Market Surveillance ist die Fortsetzung und Aktualisierung der klinischen Bewertung. Nachdem das Produkt auf dem Markt ist, wird die Funktionsweise des Produkts weiterhin beobachtet und die Daten zur Produktverwendung gesammelt, analysiert und aktualisiert.

Bezüglich der Anforderungen in der klinischen Bewertung, klinischen Prüfung und der Post Market Surveillance gibt es viele Überschneidungen. Warum erstellen Sie dann nicht gleich eine Art "Handbuch" für die ohnehin vorzunehmende klinische Bewertung, auf das Sie auch für PMS oder im Falle des Falls auch für die klinische Prüfung zurückgreifen können? In der folgenden Tabelle haben wir Ihnen eine Übersicht erstellt, aus der die Überschneidungen hervorgehen:
| Handbuch für klinische Bewertung, klinische Prüfung und PMS | |||
| A. Klinische Bewertung | B. Klinische Prüfung | C. Post Market Surveillance | |
| Ziel | Untermauern der Zweckbestimmung, Sicherheits- und Leistungsanforderungen mit vorhandenen klinischen Daten | Untermauern der Zweckbestimmung, Sicherheits- und Leistungsanforderungen mit neuen klinischen Daten | Aktualisierung der Zweckbestimmung, Sicherheits- und Leistungsanforderungen mit beobachteten Daten |
| Methode | Literaturreview / Metaanalyse | Randomized Controlled Trial (RCT) | Vervollständigung des Literaturreview / Metaanalyse |
| Outcome | angestrebter klinischer Nutzen | angestrebter klinischer Nutzen | angestrebter klinischer Nutzen |
| Zielpopulation | Spezifizierung der Zielgruppe | Spezifizierung der Zielgruppe | Spezifizierung der Zielgruppe |
| Fallzahlplanung | Bewertung methodischer Qualität, Subgruppen und Vergleichbarkeit statistischer Kennwerte | Poweranalyse zur Ermittlung geeigneter Stichprobengröße | Bewertung methodischer Qualität der eingegangenen Beobachtungsdaten |
| Ergebnis | Zusammenfassung der Ergebnisse | Datenauswertung | Aktualisierung der Ergebnisse |
| Diskussion | Bewertung der relevanten Daten hinsichtlich Vollständigkeit für die klinische Bewertung | Interpretation der Ergebnisse und Bewertung der relevanten Daten hinsichtlich Vollständigkeit für die klinische Bewertung | Interpretation der Gesamtergebnisse und Bewertung hinsichtlich des Einhaltens der Zweckbestimmung, Sicherheits- und Leistungsanforderungen |
Wie Sie in 7 Schritten wichtige Anforderungen der Kapitel 6 und 7 der MDR umsetzen:
1. Verschaffen Sie sich einen Überblick über unternehmensinterne Informationen - hierfür sollten Sie sich intern mit allen involvierten Bereichen abstimmen, z.B. mit Market Access, Medical und Regulatory Affairs. Fassen Sie alle internen Daten und Datenlücken zusammen.
2. Gleichen Sie diese Informationen mit unternehmensexternen Informationen ab, indem Sie eine systematische Literaturrecherche durchführen. Die Bewertung der externen Informationen generieren Sie, indem Sie ein systematisches Review oder eine Metaanalyse erstellen. Beachten Sie hierbei, dass die Aussagekraft der systematischen Übersichten und der Metaanalysen stark von der Systematik der Recherche und Kategorisierung der Publikationen abhängt. Vermindern Sie systematische Verzerrungen, indem Sie beispielsweise die Risk of Bias Analyse des Cochrane Tools verwenden. Achten Sie darauf statistische Kennwerte wie Effektstärken und Korrelationskoeffizienten zu kommunizieren, um einen höheren Objektivitätsgrad zu erreichen. Halten Sie Ihr Ergebnis auch in einem Prisma-Schema fest.
3. Prüfen Sie, ob es zu Ihrem Produkt interne oder externe Äquivalenzstudien gibt. Sind Äquivalenzstudien vorhanden, halten Sie deren Ergebnisse im Rahmen indirekter Vergleiche fest.
4. Legen Sie eine Tabelle an, in der die Informationen aus 1, 2 und 3 zusammengefasst werden. Versehen Sie diese mit einem Evidenzgrad und leiten hieraus den klinischen Nutzen für den Patienten ab, abgestuft von patientenrelevanten Nutzen hin zu Surrogatparametern. Sie wissen nun wie die vorhandene Evidenz einzuordnen ist, ob eine Evidenzlücke vorliegt und welche messbaren, vorhandenen klinischen Informationen sie kommunizieren können. Spätestens hier wissen Sie, ob auch noch klinische Prüfungen erforderlich sind.
5. Sollte noch eine klinische Prüfung erforderlich sein, dann erstellen Sie eine Checkliste der weiteren to-dos. Dafür ist der Anhang XV, Kapitel II hilfreich. Achten Sie auf die wissenschaftliche und methodische Qualität der Prüfungen, denn RCTs sind häufig mit hohen Kosten verbunden und zeitaufwendig. Legen Sie hier daher a priori Gütekriterien fest (z.B. Poweranalysen für erforderliche Stichprobengröße) und das Umgehen mit möglichen Änderungen in der klinischen Prüfung, z.B. vorzeitiger Abbruch oder festgestellte Produktmängel.
6. Nutzen Sie praktische Hilfen zur Überwachung nach dem Inverkehrbringen, beispielsweise indem Sie mittels des kostenfreien Tools Mendeley eine Fachliteraturdatenbank anlegen und mittels Alerts ständig aktualisieren. Auch ein Frühwarnsystem zur Erfassung aller Vorkommnisse und Nebenwirkungen, z.B. Rückmeldungen von Anwendern und Händlern, werden Sie installiert haben. Überführen Sie diese Informationen unmittelbar in Ihr PMS-System. Da Sie mindestens einmal im Jahr (Risikoklassen IIb und III) bzw. alle zwei Jahre (Risikoklassen IIa) eine bewertende Zusammenfassung auf Basis aller Änderungen vornehmen müssen, hilft eine solche kontinuierliche Dokumentation von Informationen auf angekündigte und nicht angekündigte Audits immer vorbereitet zu sein.
7. Fassen Sie alles in einem Dokument zusammen (Ihrem Handbuch für die klinische Bewertung).
Der Übergang zur MDR bedeutet für Sie als Hersteller einen nicht zu unterschätzenden zeitlichen Aufwand, insbesondere im Hinblick auf die klinische Bewertung und Marktüberwachung. Schaffen Sie daher frühzeitig Standards und Synergien mit denen Sie die drei Aufgaben klinische Bewertung, klinische Prüfung und Post Market Surveillance analog angehen können. Integrieren Sie dieses Vorgehen in ihr Qualitätsmanagementsystem, dann können Sie dieses zeitgleich auch für die Zertifizierung dieses Systems nutzen.

Tino Schubert hat am 21.03.18 beim BVMed einen Vortrag zum Status Quo der integrierten Versorgung gehalten. Unter anderem war auch das Thema Evidenzgenerierung und die Übertragung auf verschiedene Verwendungszwecke Gegenstand der Veranstaltung – fordern Sie die Vortragsunterlagen kostenfrei hier an.
Sprechen Sie uns an – wir zeigen Ihnen an Beispielen und unserer praktischen Erfahrung welche Instrumente Sie für klinische Bewertung und ein effizientes PMS System verwenden können. Wir freuen uns auf den Austausch mit Ihnen.
Hintergrundinformationen zur Veranstaltung finden Sie hier.
Parallel zur Zulassung Ihres Produkts im Rahmen der MDD oder MDR sollten Sie sich auch über die passende Erstattungsgrundlage in der gesetzlichen Krankenversicherung und die notwendigen Schritte zu einer Erstattung Gedanken machen. Fordern Sie hierzu unser kostenloses White Paper Erstattungswege für Medizinprodukte im deutschen Gesundheitswesen an (hier kostenfrei herunterladen).
About
LinkCare GmbH
Schwieberdinger Straße 52
71636 Ludwigsburg
Deutschland





